 Richard A. Bareš
Richard A. Bareš
1. Struktura materiálů jako hlavní faktor určující jejich vlastnosti
Hlavní vlastnosti tuhých materiálů závisí na charakteru jejich struktury. Obecně platí, že čím je struktura disperzního materiálu hustší a stejnorodější, tím vyšší je jeho pevnost, vodotěsnost, mrazuvzdornost a odolnost proti opotřebení i korozi.
Vlastnosti ztvrdnutých polymermalt (a tím i polymerbetonů), jež jsou typickým disperzním kompozitním materiálem s velmi rozvinutým povrchem mezi fázemi, závisí především na jejich pórovité struktuře a struktuře pevné fáze. Je přirozené, že charakter pórovité struktury se nejzřetelněji projevuje při zkouškách vzorků na vodotěsnost a mrazuvzdornost .
Při zkouškách na mrazuvzdornost, spočívajících v nasycení vzorků ztvrdnuté malty vodou a v následném jejich střídavém zmrazování a rozmrazování se mikrokapiláry nenaplňují vodou úpině. Proto tyto mikrokapiláry slouží jako by rezervní objem, kam se působením sil vznikajících při zvyšování objemu vody v mikrokapilárach v důsledku zmrazování (při tlacích do 200 MPa a při zvětšení objemu do 9 % ) tato voda přemísťuje.
Významnou roli hraje kohezní pevnost v oblastech kontaktu polymerních novotvarů ztvrdnuté kaše a to jak navzájem mezi sebou, tak i s plnidlem obsaženým
v betonové směsi. Kohezní pevnost polymermalty je charakterizována schopností vzdorovat různým mechanickým vlivům (tlak, ohyb, náraz, otěr).
Podstatný význam pro vytváření polymermaltových povrchů má adheze tohoto povrchu k základnímu materiálu (podkladu). V čerstvě položené maltě nanesené na zatvrdnutém podkladu vznikají v průběhu tvrdnutí vnitřní smršťovací napětí, jejichž veličina může někdy i značně převyšovat sily adheze, což vede současně ke snížení hodnoty adheze a následně k odloupnutí povrchu od podkladu.
Přitom v samotném pokrytí mohou vznikat trhliny jako důsledek převýšeni veličiny vnitřních smršťovacích napětí nad kohezními silami zatvrdnuté polymermalty, což je často taktéž příčinou snížení vodotěsnosti, mrazuvzdornosti a odolnosti pokrytí vůči korozi v důsledku koncentrace napětí v izolovaném vrcholu trhliny.
2. Pojem defektů struktury a kriteria optimální struktury polymermalt
Nejrůznorodější kompozitní disperzní materiály, včetně zatvrdnutých polymermaltmalt, podle mechanických charakteristik, hlavně z hlediska pevnosti a pružně — viskózně — plastických vlastností, jsou z pohledu fyziky pevných látek skutečnými pevnými látkami.
Na rozdíl od ideálních bezdefektních pevných látek, jejichž pevnost odpovídá pevnosti meziatomových vazeb a destrukce při kritickém napětí je doprovázena nevratnou disociací, v reálných pevných látkách defekty a nehomogennosti struktury určují jiný charakter destrukce, spočívající v rozpadu reálného tělesa na části (bloky) při napětích o několik řádů nižších než je teoretická pevnost ideálního tělesa.
Protože prozatím jsou v současné technice a technologii používány materiály charakterizované vlastnostmi reálného pevného tělesa, je základním úkolem celková optimalizace struktury a vlastností reálných disperzních materiálů. To znamená, že v první řadě je nezbytné odstranit takové druhy defektů a nehomogenností struktury materiálu, které jsou bezprostředními zdroji poklesu pevnosti, destrukce (rozpadu) nebo zhoršení jeho jiných vlastností.
Analýza řady prací z oboru teorie pevnosti a destrukce (rozpadu) pevných látek dovoluje učinit předpoklad o možnosti klasifikace nejvíce rozšířených typů defektů a nehomogenností reálných kompozitních disperzních materiálů.
Podstatou této klasifikace je rozdělení defektů a nehomogenností podle třech příznaků (Tab. 1): měřítka (rozměru), druhu (tvaru ve fyzikálně-chemickém smyslu) a podstatě (původu, vzniku).
Ve shodě s představami fyziky a chemie disperzních systémů a jejich části a fyzikálně chemické mechaniky dispersních struktur pevných látek, spočívá všeobecný základ optimalizace struktury a vlastností dispersních materiálů v odstranění rozměrově největších defektů a nehomogenností I. druhu, nezávisle na jejich vzhledu a původu, nakolik právě ony jsou bezprostředním zdrojem destrukce (rozpadu), fyzikálně chemické nestability a nehomogennosti materiálu.
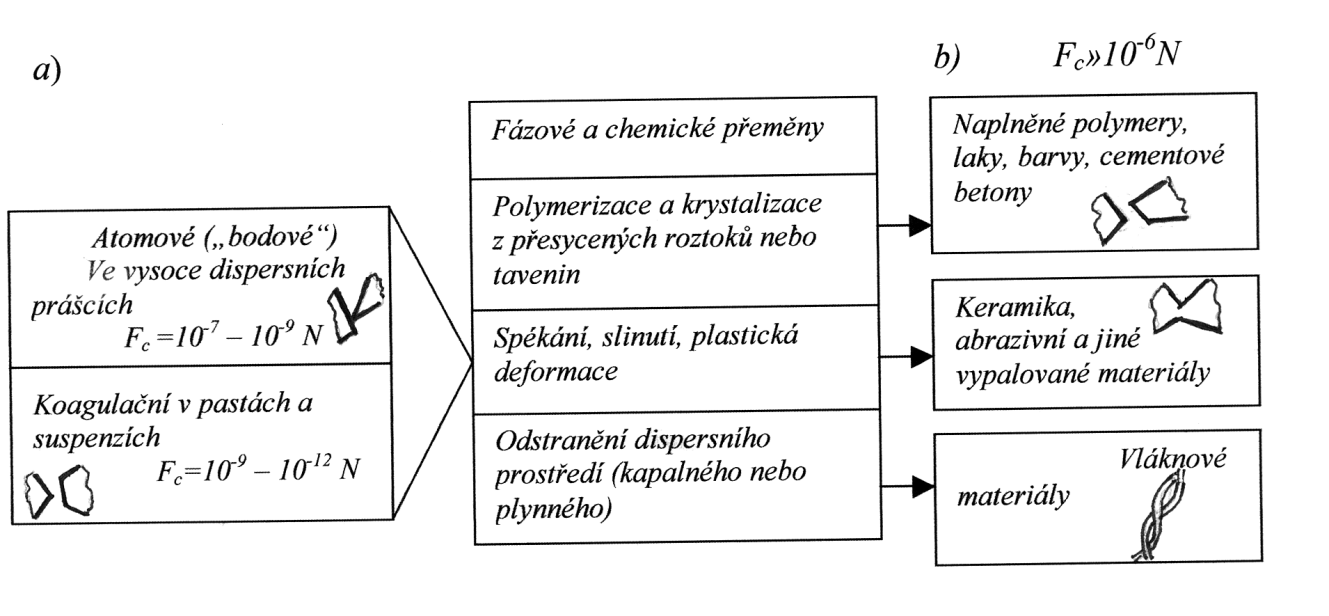
V případě reálných kompozitních materiálů mezní úroveň optimalizace struktury je vymezena možností zmenšení všech typů defektů a nehomogenností až do rozměrů, které odpovídají mozaikovému bloku struktury pevného tělesa (10-7 – 10-8 m), tj. do úrovně nehomogenností a defektů III. druhu (Tab. 1).
Při realizaci této podmínky je nutno vycházet z představy, že kompozitní dispersní materiály, včetně těch na polymerním základě, jsou z velké části vytvořeny jako výsledek distribuce rozličných pevných dispersních fází navzájem mezi sebou v kombinaci s kapalným prostředím a s následným ztvrdnutím struktury v důsledku fázových přeměn. Zpravidla takové materiály obsahují taktéž určité množství pórů, jež jsou důležitým prvkem jejich struktury.
Tabulka 1.
Zobecněná klasifikace základních typů defektů a nehomogenností reálných kompozitních dispersních materiálů
| Klasifikace podle rozměrů | Klasifikace podle tvaru | Klasifikace podle původu |
|---|---|---|
| 1. Nehomogennosti a defekty I. druhu (velké póry), Ø ≥ 10-5m | 1. Fyzikální nehomogennosti vytvářející strukturu dispersních fází, podle rozměru, tvaru a distribuce v objemu struktury | 1. Nehomogennosti a defekty spojené s mechanismem vzniku struktury a s jeho podstatou (přítomnost pórů, vzniklých smršťováním během tvrdnutí jako důsledek polymerace nebo defektů v polymerních systémech, spojených s mechanismem vylučování nové polymerní fáze a s tvrdnutím při odstranění rozpouštědla) |
| 2. Nehomogennosti a defekty II. druhu (mikrotrhliny), Ø =10-5 – 10-7 m | 2. Fyzikálně-chemická nehomogennost (termodynamická nestabilita fázových kontaktů) | 2. Nehomogennosti a defekty vznikající v důsledku nedokonalostí technologického procesu |
| 3. Nehomogennosti a defekty III. druhu (ultramikropóry, dislokace v krystalech), Ø =10-7– 10-10 m | 3. Nehomogennost chemického složení dispersních fází a části povrchu částic, jejich lyofilně lyofobní mozaikovost, nehomogennost stavu napětí v objemu struktury | 3. Největší nehomogennosti a defekty (I. druhu), vzniklé v důsledku hrubého porušení používané technologie (zanedbání péče během tvrdnutí betonu nebo malty, přítomnost hliněných tenkých vrstev a znečištění na povrchu kameniva, porušení režimů tepelného zpracování atd.) |
Z výše uvedeného výkladu plyne, že pro zmenšení rozměrů nehomogenností a defektů jako základní podmínky optimalizace struktury a vlastností dispersních materiálů je nezbytné:
- zvýšit stupeň disperse pevné fáze výchozích složek tím, že se odpovídajícím způsobem zmenší rozměr částic na hodnoty 10-7 — 10-8 m;
- zmenšit tloušťku mezivrstvy, která slepuje částice dispersního prostředí v počátečním stadiu na hodnoty 10-7 — 10-8 m, čehož se dá dosáhnout zvýšením objemového obsahu pevné fáze v dispersním prostředí;
- zmenšit na minimum nebo úpině vyloučit pórovitost tím, že se převedou zbývající póry na mikro- nebo ultramikropóry.
Nicméně výše uvedené podmínky jsou sice nutnými, avšak nejsou postačujícími k získání materiálů s optimalizovanou strukturou. Jedná se o to, že zvýšení dispersní schopnosti S a objemového obsahu φ pevné fáze v dispersním prostředí, nutných k realizaci těchto podmínek, prudce zvyšuje agregovatelnost soustavy, t. j. vede ke zvýšení pravděpodobnosti vzniku fyzikální nehomogennosti a defektnosti struktury (Tab. 1), a to v míře tím větší, čím větší je S a φ. Proto úkol technologie přípravy dispersních materiálů spočívá v tom, aby při optimalizované dispersní schopnosti Sopt a objemovém obsahu pevné fáze φopt, byla minimalizována nebo úplně vyloučena záporná role „vrozených" defektů a nehomogenností, vznikajících v důsledku nedokonalosti technologického procesu nebo nedodržení daných technologických parametrů. Současně je nezbytné minimalizovat fyzikálně chemickou nehomogennost a nestabilitu struktury a zmenšit hladinu napětí II. druhu.
Z uvedeného vyplývá, že při přípravě dispersních materiálů je základním bezprostředním úkolem technologického procesu dosáhnout mezní homogennosti distribuce dispersní pevné fáze při jejím optimalizovaném obsahu v dispersním prostředí v počátečním stadiu technologického procesu a zachování dosažené homogennosti a vysoké dispersity částic po ukončení fázových přeměn, tj. po vytvoření pevné (zatvrdnuté) struktury materiálu.
3. Způsoby řízení vzniku struktury a procesu vytváření struktury kompozitních materiálů
V řadě moderních kompozitních materiálů mají, z hlediska rozmanitosti a objemu aplikací, nejvýznamnější místo dispersní materiály, vysoce naplněné pevnou fází, vytvářené z dispersních systémů jako výsledek realizovaných chemických a fázových transformací.
Polymermalty jsou typickým příkladem kompozitních dispersních materiálů tohoto typu. Charakteristická zvláštnost těchto dispersních materiálů naplněných pevnou fází spočívá v tom, že v počátečním stádiu jejich přípravy z dispersních systémů samovolně vznikají termodynamicky stabilní prostorové struktury (z částic pevné fáze s reverzibilně se rozpadajícími koagulačními kontakty mezi těmito částicemi). Tyto prostorové struktury vznikají v důsledku silně rozvinutého povrchu rozhraní mezi fázemi a vysoké koncentrace dispersních fází v kapalném prostředí. Pak podle míry uskutečňování chemických a fázových přeměn (polymerace v nejširším smyslu)) na pozadí struktur s reverzibilními kontakty vznikají a stávají se převažujícími struktury s pevnými skutečnými a nereverzibilními fázovými kontakty, které však určují strukturně mechanické vlastnosti cementových se rozpadajícími kontakty, které určují strukturně mechanické vlastnosti zatvrdlého materiálu.
Tímto způsobem, z rozmanitosti dispersních systémů, v nichž částice pevné fáze vytvářejí třírozměrné prostorové mřížky, je možné vyčlenit tři základní typy struktur v souladu s typem kontaktů mezi částicemi.
Prvnímu typu kontaktů odpovídají koagulační struktury tvořené částicemi pevné fáze které jsou rozděleny rovnovážnými mezivrstvami kapalného dispersního prostředí. Druhý typ struktur je tvořen bezprostředními bodovými „suchými" atomovými kontakty mezi částicemi. Struktury s koagulačními a atomovými kontakty převažují v počátečných etapách technologie přípravy většiny dispersních materiálů v procesu dávkování jednotlivých komponent, jejich promíchávání, nanášení hotových směsí, formování výrobků nebo zhutňování. Nejpevnější prostorové krystalizačně-kondenzační struktury jsou vytvářeny skutečnými fázovými kontakty třetího typu, které jsou charakteristické pro zatvrdnuté struktury dispersních materiálů (Obr. 1).
Koagulační struktury jsou jedním ze základních typů struktur. Z hlediska jejich interakce se vyznačují poměrně slabými kontakty mezi částicemi. Pevnost těchto kontaktů je určena van der Waalsovými molekulárními silami koheze podél dílčích lyofobních částí makromozaikového povrchu částic přes nejtenčí mezivrstvy dispersního prostředí, jejichž ustálená tloušťka odpovídá minimální hodnotě volné povrchové energie soustavy.
Podle výsledků teoretického výpočtu a na základě výsledků experimentálního výzkumu u koagulačních struktur dosahuje síla interakce částic dispersní fáze vztažená na jeden kontakt středních hodnot 10-9- 10-12 N.
Nicméně, skutečná pevnost kontaktu závisí do značné míry na podmínkách jeho vytváření, které určují ustálenou rovnovážnou tloušťku mezivrstvy kapalné fáze mezi částicemi. V průběhu přibližování částic v oblasti působení van der Waalsových sil tyto částice překonávají energetickou bariéru v strukturované kapalině nebo odpudivé elektrostatické síly, vznikající v důsledku vytvoření dvojitých adsorpčních vrstev.
V souladu s těmito představami částice v koagulační struktuře se mohou ustálit ve vzdálenosti těsné (hmin≈ 10-9 m) nebo řídké (h2 = 10-7 m) koagulace, což určuje rozdíl přibližně dva řády v energiích a síle vazby mezi nimi Pevnost a vazebná energie koagulačních kontaktů prudce klesají při pokrytí povrchu částic jedinou vrstvou povrchově aktivní látky (PAL). V tomto případě polární skupina molekuly PAL je adsorbována bezprostředně na povrchu pevné fáze (je-li tento povrch hydrofilní), a uhlovodíkový řetězec je obrácen navenek. Při adsorpci PAL se oddalují částice do vzdálenosti minimálně dvou molekulárních vrstev , přičemž současně stíní energeticky nejaktivnější dílčí části makromozaikového povrchu částic.
Interakce mezi částicemi se při tom uskutečňuje například prostřednictvím uhlovodíkových metylových skupin CH3 s minimální pevností vazby. Nutnou podmínkou vytváření skutečných koagulačních struktur je přítomnost částic pevné fáze, majících koloidní rozměry φi (10-9— 10-7) m v celém souboru částic pevné fáze a schopných vykonávat Brownův pohyb.
V reálných soustavách jsou zpravidla obsaženy frakce neisomerních částic, jejichž střední rozměr může být jak větší, tak i menší než je výše uvedený rozměr. Nicméně, je-li v soustavě i jen celkem nevelký zlomek počtu částic o rozměrech vyhovujících podmínce φ « φi, je nutné vzít v úvahu kontaktní interakce mezi nimi. Prakticky všechny typy dvoufázových a třífázových soustav obsahují částice pevné fáze, pro které platí φ « φi . Tento podíl vysoce dispersních částic o koloidních rozměrech může dosahovat pouhých několik percent z celkového počtu částic pevné fáze. Tím, že jsou rozloženy v celém dispersním objemu, vytvářejí v souhrnu s většími částicemi prostorový třírozměrný skelet, skládající se z řetězců nebo agregátů. Pravděpodobnost a rychlost vytváření struktur je tím vyšší, čim vyšší je jejich stupeň disperse ( a tudíž schopnost účastnit se v tepelném Brownově pohybu) a čím silněji je vyjádřena anisometrie nebo lyofobně- lyofilní mozaikovost.
Přítomnost mezivrstev dispersního prostředí mezi částicemi koloidních rozměrů určuje nejdůležitější zvláštnosti koagulačních struktur tohoto druhu: — schopnost úplné tixotropní regenerace v časovém úseku po destrukci (rozpadu) a existence rychlé i zpožděné elasticity. Posledně uvedená vlastnost koagulačních struktur je spojována s orientačními efekty neisomerních částic dispersní fáze v dispersním prostředí ve směru smykové (translační) sily.
Koagulační struktury mají ve srovnání s jinými typy dispersních struktur následující zvláštnosti:
- fixaci částic pevné fáze v kapalním dispersním prostředí ve vzdálenosti těsné nebo řídké koagulace;
- tixotropní reverzibilitu v důsledku přítomnosti částic schopných konat Brownův pohyb;
- projevení rychlé nebo zpomalené pružnosti, tečení a nízké viskozity
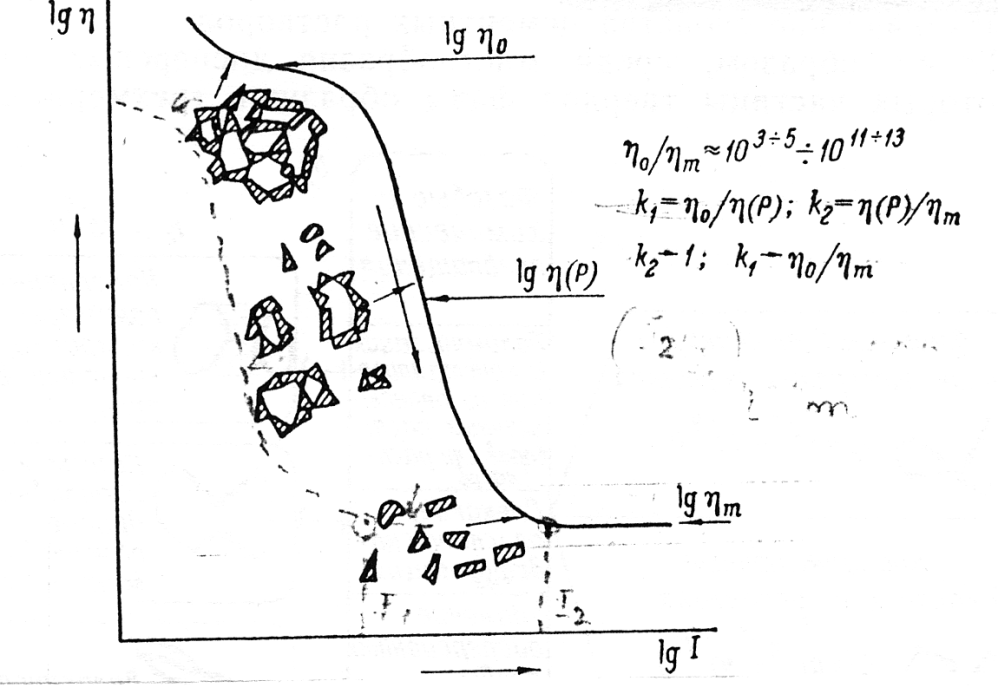
Současně s tímto se koagulační struktury vyznačují ostře vyjádřenou závislostí strukturně- mechanických charakteristik na působení fyzikálně-chemických a mechanických činitelů. Příklad výjimečné citlivosti strukturně mechanických vlastností dvoufázových tixotropních koagulačních struktur ve vztahu k mechanickým vlivům v průběhu transportu hmoty je závislost rovnovážné efektivní viskozity η(P) při spojité smykové deformaci soustavy na rychlosti deformace έ = dη/dt, nebo na smykovém napětí P. Úroveň η(P) odpovídá definovanému stupni destrukce (rozpadu) tří rozměrového strukturního skeletu v podmínkách deformace systému s danou rychlostí smyku.
V tixotropních koagulačních dvoufázových strukturách může rozsah změn η (P) =f(έ P) dosahovat až 9 —11 dekadických řádů. Proto úplná reologická křivka tečení takových soustav (Obr. 2), která vyjadřuje funkční závislost mezi úrovní η (P) a rychlostí smyku ε (smykovým napětím P), je nejdůležitější charakteristikou koagulační struktury v počátečních etapách procesu vzniku dispersních materiálů, zejména polymermalt a polymerbetonů.
Úpiná reologická křivka může být získána pro takové koagulační struktury, u nichž je zachována schopnost úplné tixotropní regenerace po procesu destrukce (rozpadu). Nicméně, úplná tixotropní reverzibilita v koagulačních strukturách se zachovává pouze do určité konkrétní kombinace hodnot objemového obsahu (φ) a dispersní schopnosti částic pevné fáze S v kapalném dispersním prostředí, které jsou specifické pro každý konkrétní případ. Při překročení φ a S těchto daných konkrétních hodnot se zpočátku ztrácí schopnost tixotropní regenerace v celém objemu deformované soustavy a s postupným následným relativním poklesem obsahu kapalné fáze v dispersní soustavě tato ztrácí své vysoce elastické a následně i plastické vlastnosti.
Nejobecnější pohled na specifiku dispersních soustav s kontakty koagulačního typu ukazuje, že změny jejich základních strukturních parametrů:
- dispersní schopnosti
- objemového obsahu dispersní fáze v kapalném prostředí
- zavedení přísad povrchově aktivních látek (PAL)
dovoluje do značné míry (v mezních případech až o více než o 10 řádů) změnit strukturně mechanické vlastnosti koagulačních struktur. Tyto změny jsou doprovázeny vznikem nebo vymizením specifických vlastností, charakteristických pouze pro koagulační struktury:
- meze pevnosti (při existenci struktury pevné fáze — při absenci kapalné),
- tixotropní reverzibility vlastností,
- plasticity a elasticity
Pokles obsahu kapalného dispersního prostředí v těchto strukturách je doprovázen přechodem od dvoufázových k třífázovým soustavám, při současném vytváření kapilárních menisků a v důsledku toho i odpovídající změnou pevnostních charakteristik struktury.
Nakonec, při úplném odstranění kapalného dispersního prostředí v těchto strukturách je možné pozorovat přechod k hutným a pevným strukturám, v nichž jsou částice vázány jedna s druhou prostřednictvím přímých bodových kontaktů s plochou, která je souměřitelná s rozměry několika atomů nebo buňky krystalické mřížky.
Ve sledované řadě přechodových koagulačních struktur se nejvíce přibližují ke kondenzačně krystalizačnímu typu s pevnými fázovými kontakty mezi částicemi struktury s bodovými kontakty. Přeměna koagulačních struktur s bodovými kontakty na kondenzačně krystalizační s fázovými kontakty je v některých případech možná a to jako výsledek krystalizace z přesycených roztoků (např. cementová malta).
Hlavní rysy kondenzačně krystalizačních struktur:
- vysoká pevnost v porovnání s koagulačními strukturami určovaná vysokou pevností samotných fázových kontaktů mezi částicemi;
- nevratný charakter destrukce (rozpadu) - jasně vyjádřená pružnost i křehkost a velmi nízká plasticita,
- přítomnost vnitřních napětí, vznikajících v samotném procesu tvorby fázových kontaktů.
Mezi různými typy kondenzačně krystalizačních struktur zajímají významné místo krystalizační struktury s hydratačním tvrdnutím.
Zároveň se všeobecnými, výše zmíněnými příznaky kondenzačně krystalizačních struktur, krystalizační struktury s hydratačním tvrdnutím mají řadu specifických zvláštností, spojených se samotným mechanismem vytváření krystalické prorostliny v procesu hydratace vysoce dispersní výchozí fáze.
Bylo potvrzeno, že nutnou podmínkou vzniku struktur s fázovými kontakty v závěrečních etapách přípravy dispersních materiálů je, aby v soustavách na počátku procesu docházelo k vytváření struktur s reverzibilními, pokud se jedná o pevnost, koagulačními kontakty.
Proto je důležité následně stanovit, jaké jsou podmínky vzniku takových reversibilních, vzhledem k pevnosti, dispersních struktur. Tyto podmínky se v podstatě redukují na stanovení
- za prvé kritického rozměru částic φc, počínaje kterým jsou kohezní sily mezi částicemi souměřitelné s jejich hmotností,
- a za druhé hodnoty kritické koncentrace ξc1 dispersní fáze v kapalném prostředí, počínaje kterou mohou v dispersní soustavě vzniknout prostorové struktury.
Kritický rozměr částic φc je možné určit za předpokladu, že účinek vnějších sil na částice, vytvářející dispersní soustavu, v nejobecnějším a nejjednodušším případě vyvolaný zrychlením v důsledku působení gravitační síly, je vyvážen molekulárními silami koheze mezi nimi

kde Fc. je sila koheze v oblasti kontakt.
Tabulka 2.
Hodnoty kritického rozměru částic φc v závislosti na vzdálenosti H mezi nimi
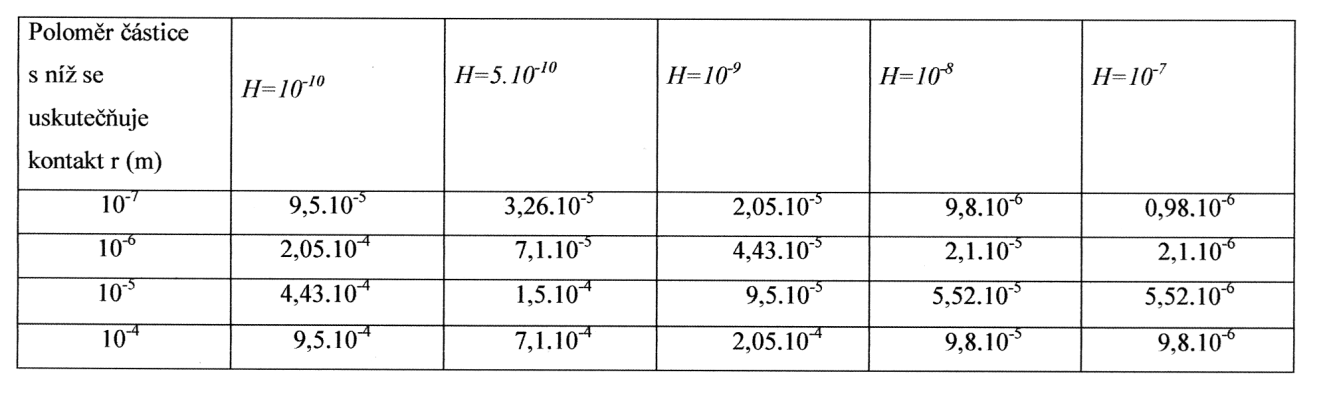
Vycházeje z této podmínky je možné na základě teorie molekulárních interakcí kondenzovaných fází stanovit kritické rozměry částic φc, pro struktury s bezprostředními atomovými (2) a koagulačními kontakty při fixaci částic v struktuře v polohách blízkého energetického minima (3) a pro řídkou koagulaci (4)v posledním případě s respektováním elektromagnetického zpoždění dispersních sil):
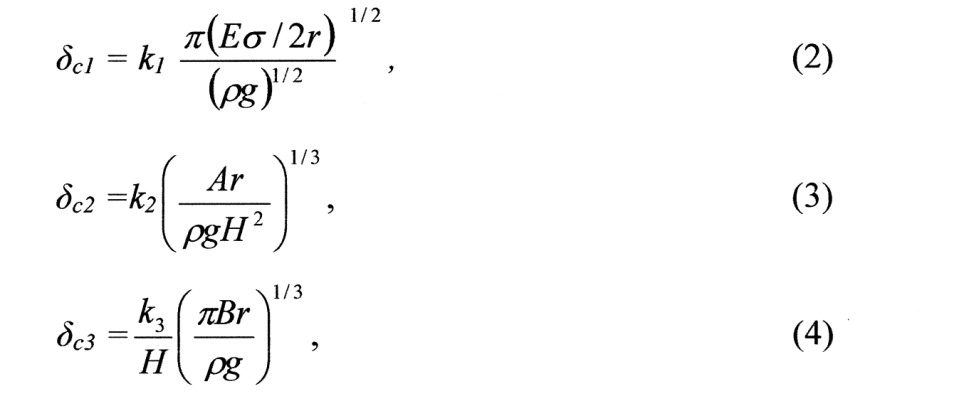
kde ρ je hustota kontaktujících se částic, E modul pružnosti, σ povrchové napětí pevné látky, r poloměr částice s níž dochází ke kontaktu, A Hamakerova konstanta, B konstanta molekulární interakce kondenzovaných fází s respektováním elektromagnetického zpoždění dispersních sil, k1, k2, k3 bezrozměrné konstantní koeficienty beroucí v úvahu zakřivení povrchu kontaktujících se částic.
Absolutní hodnoty kritických rozměrů pro analyzované případy (jako příklad byly použity křemenné částice) jsou uvedeny v tabulce 2.
Za pozornost stojí fakt, že hodnota φc o několik řádů převyšuje rozměry koloidních částic, přičemž dosahuje v řadě případů desítky a dokonce i stovky um. To znamená, že
v dispersní soustavě s takovými dostatečně velkými částicemi je možné vytváření prostorové struktury. Současně náchylnost k samovolnému vytváření struktury, která se dá charakterizovat veličinou κ =Fc/P (P — částice), s poklesem rozměru částic rychle roste (Obr. 3), minimálně v oblasti kde φi < φ < φc ( φi je rozměr koloidních částic). Stanovíme-li v obecném tvaru hodnoty φc , můžeme odhadnout hodnoty kritické koncentrace vytváření struktury ξc1 .
Hodnota ξc1 významně závisí na tvaru částic dispersních fází ( stupni anisometrie), povaze jejich povrchu, podmínkách interakce částice s prostředím atd. Skutečně, hodnota φci se může měnit ve velmi širokém rozsahu a to od setin a desetin procenta pro případ vodních dispersí V205 celulózových nebo azbestových vláken, do několika procent pro případ vodních dispersí sodíkového bentonitu a až po desítky procent pro případ vápníkového bentonitu.
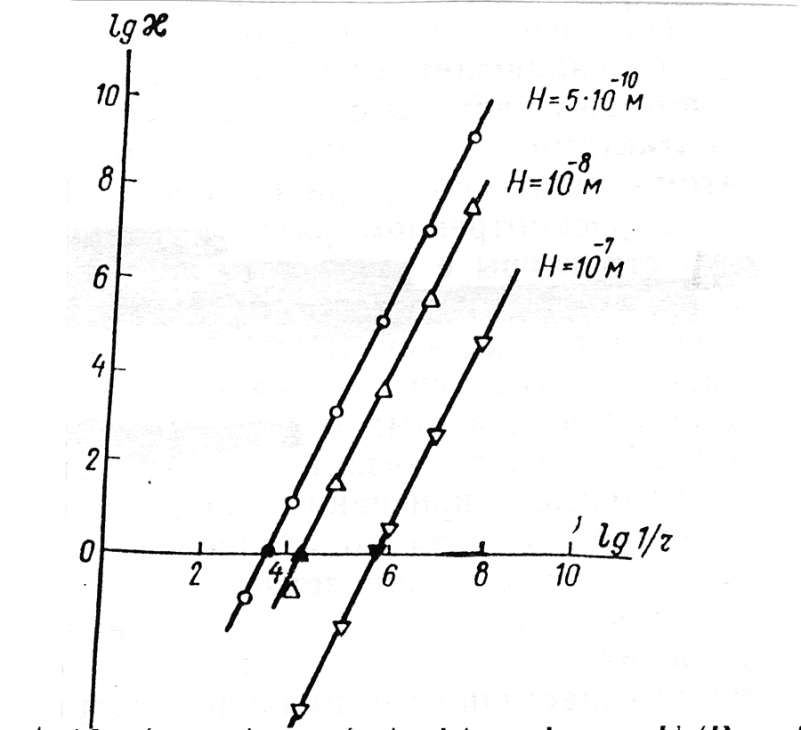
V případě dostatečně velkých (avšak současně vyhovujících podmínce Ø < Øc) isometrických částic, může hodnota φc1 dosahovat několik desítek procent, například pro vodní disperse cementu. Zmenšení rozměru isometrických částic je přirozeně doprovázeno podstatným poklesem φc1 , což je charakteristické například pro dispersi aerosilu v epoxidovém pojivu.
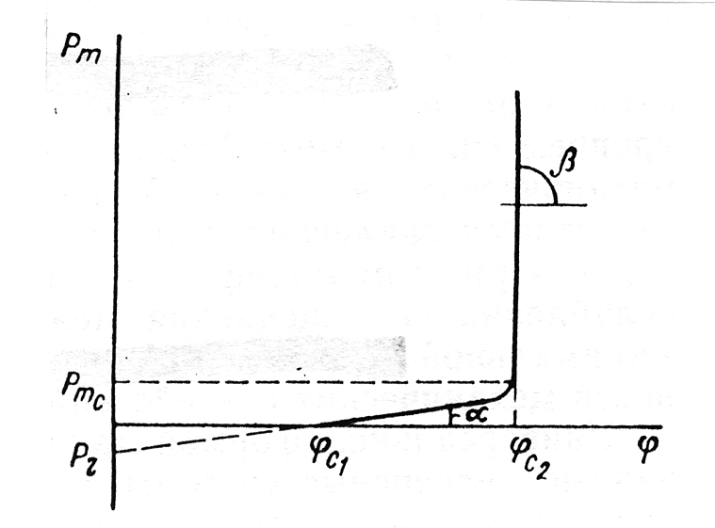
Současně s tím stanovení hodnoty kritické koncentrace dispersní fáze v kapalném nebo plynném prostředí je možné na základě teorie pevnosti pórovitých struktur. Zjištění mezního napětí smyku v dispersní soustavě Pm > 0 je možné interpretovat jako počáteční kritickou koncentraci odpovídající vzniku strukturní mřížky. Podle je možné veličinu Pm, stanovit ze vztahu (5)

kde α je koeficient respektující mikrogeometrii struktury (koordinační číslo), n počet kontaktů v objemové jednotce struktury, f(φ) funkce, charakterizující závislost pevnosti na objemové koncentraci dispergované fáze v prostředí a Ø střední charakteristický rozměr částic dispersní fáze.
Graficky může být vztah (5) znázorněn závislostí Pm, na (Obr. 4). Je důležité poznamenat, že s růstem φc > φc1 vzrůstá Pm lineárně až do φc1≤ φc2 a v úzkém rozsahu zvyšování φc1> φc2 pozorujeme prudký nárůst Pm. Veličiny tgα, tgβ, Pm, φc1 a φc2 mohou být považovány za charakteristiky koncentrační závislosti Pm —φ dispergované fáze v dispersním prostředí pro danou soustavu. Je důležité zdůraznit, že N.ah (5) platí pro pórovité struktury, prostředí pro danou soustavu. Je důležité zdůraznit, že vztah (5) platí pro pórovité struktury, jak s reverzibilnírni vzhledem k pevnosti, tak i nevratně se rozpadajícími fázovými kontakty. To znamená, že přirozený způsob zvýšení základní charakteristiky dispersních materiálů, jejichž struktura je určena fázovými kontakty mezi částicemi, jejich pevností, spočívá v kombinaci zvýšení pevnosti kontaktů a nárůstu jejích počtu n, tj. počtu částic v objemové jednotce. Následně je růst n dosahován kombinací růstu dispersní schopnosti v objemové jednotce. Následně je růst n dosahován kombinací růstu dispersní schopnosti částic pevné fáze S (odpovídajícího zmenšení (5) Ø a jejich koncentrace φ, zejména při přechodu do oblasti φ≥ φc2.
Avšak do té míry, do jaké vztah (5) reflektuje nárůst pevnosti struktury dispersních materiálů s růstem φ a S, tentýž vztah současně reflektuje spojitý růst pevnosti Pm, viskozity η (obzvláště prudký je tento růst v oblasti, v níž je φ> φc2) a tudíž ztrátu fluidity a vysoké pohyblivosti dispersních soustav s reversibilními, z hlediska pevnosti, kontakty, z nichž vznikají struktury dispersních materiálů s nereversibilně se rozpadajícími fázovými kontakty.
Odpovídajícím způsobem, v důsledku ztráty fluidity a vysoké pohyblivosti, prudce klesají možnosti řízení strukturně reologických vlastností dispersních soustav při uskutečňování procesů míchání složek, formování, zahuštění atd., tj. všech těch technologických postupů založených na transportu hmoty, bez nichž by nebylo možným získání dispersních materiálů s předem danými vlastnostmi.
Proto realizace vztahu (5), tj. zvýšení pevnosti dispersních materiálů v oblasti φ> φc2 v souladu s tímto vztahem je možné pouze do takové míry, do jaké v počátečních etapách procesu vytváření struktury v době převažování reverzibilních kontaktů mezi částicemi v dispersních soustavách se dosahuje snížení pevnosti Pm, a viskozity η (P) struktury, při struktury, při zachování vysokých hodnot φ a S.
Vyřešení tohoto významného protikladu, spočívajícího v nutnosti, z jedné strany zvyšovat φ a S dispersní fáze pro získání vysoce pevných a vysoce hutných materiálů a z druhé snižovat pevnost a viskozitu dispersních soustav, z nichž jsou tyto materiály vytvářeny, tvoří hlavní úkol současné technologie dispersních materiálů. Zejména proto možnost fyzikálně chemického řízení procesů vytváření struktur a vlastností koncentrovaných dispersních soustav s reverzibilními, z hlediska pevnosti, kontakty opět určuje možnost efektivní realizace procesů transportu hmoty, jež tvoří podstatu technologie přípravy kompozitních materiálů.
Hlavní podmínka takového fyzikálně chemického řízení vlastností koncentrovaných dispersních soustav spočívá v maximálním snížení jejich efektivní viskozity a v důsledku toho dosažení jejich nejvyšší fluidity, formovatelnosti, zhušťovatelnosti při zachování relativně malého obsahu kapalného prostředí . To je možné za podmínek dosažení a udržení v čase objemového rozpadu (destrukce) těchto struktur, což znamená přerušení všech koagulačních kontaktů mezi částicemi .. Takovým způsobem, nejdůležitější charakteristika vysoce koncentrovaných dispersních soustav, která tvoří podstatu přípravy dispersních materiálů, homogennost jejich struktury, může být dosažena v podmínkách procesů transportu hmoty, který je doprovázen mezním rozpadem (destrukcí) všech koagulačních kontaktů, tj. pro případ maximální fluidity soustavy.
Tato poslední podmínka je v principu realizována při smykové deformaci dispersní soustavy rychlostí έ ≥ έ m dostačující k tomu, aby bylo dosaženo nejmenší úrovně Newtonovy viskozity ηm, například v procesu získávání úplných reologických křivek tečení plasticko-¬viskózních soustav.
Avšak v těchto podmínkách se daří mezně rozbít (destruovat) koagulační strukturu v dispersních soustavách s relativně nízkou koncentrací . Při přechodu k vysoce koncentrovaným soustavám jejich deformace, dokonce s velmi malou rychlostí smyku, jsou zpravidla doprovázeny objevením se lokálního tixotropně neregenerujícího se toku, a proto nereverzibilního, přerušení celistvosti, které vylučuje možnost dosažení v celém objemu deformované soustavy mezního rozpadu (destrukce) struktury . Základní metoda překonání této nejdůležitější překážky na cestě k dosažení nejmenší úrovně viskozity a tudíž získání homogenní struktury spočívá v následujícím.
Vzhledem k tomu, že strukturně reologické vlastnosti koncentrovaných dispersních soustav se určují vztahem mezi úrovní sil interakce částic a intenzitou vnějšího mechanického působení na strukturu, řízení těchto vlastností spočívá v optimálních změnách obou faktorů. Přitom modifikace povrchu částic, s cílem zeslabení vazebních sil mezi nimi, dovoluje dosáhnout minimální newtonovské viskozity při poklesu intenzity mechanického působení.
Univerzální formou takového působení na kondenzované dispersní soustavy jsou mechanické kmity (vibrace), které dovolují vytvořit a udržovat regulovatelný dynamický stav dispersních soustav . Tento stav je dán úrovní izotropního rozpadu (destrukce) struktury v dispersní soustavě, přitom optimálnímu dynamickému stavu odpovídá maximální fluidita, tj. nejmenší newtonovská viskozita v objemu soustavy nebo v jeho dané části. Stejný dynamický stav dispersních soustav, při uskutečnění řady často navzájem značně odlišných fyzikálně chemických procesů transportu hmoty, je určen stejnou úrovní (nebo rozsahem měření) základních reologických vlastností dispersních soustav.
Z výše uvedeného vyplývá, že superpozice objemového vibračního pole na spojitě deformovanou soustavu, s výjimkou objevení se přerušení celistvosti v takových soustavách, dovoluje principiálně dosáhnout libovolného stupně objemového rozpadu (destrukce) struktury.
Nejmenší úroveň newtonovské viskozity při vibracích se dosahuje pro měrný výkon vibračního pole daný vztahem

kde β je koeficient respektující útlum kmitů v soustavě, χ poměr amplitudy posuvu částic vzhledem k prostředí k amplitudě posuvu dispersní soustavy, αm maximální hodnota amplitudy posuvu soustavy a θm perioda relaxace odpovídající úrovni ηm, přičemž θm = ηm /Gm (kde Gm je modul pružnosti).
Uvedený vztah byl odvozen teoreticky za následujících předpokladů: výkon vibračního pole, přiváděný do dispersní soustavy, nutný k přerušení koagulačních kontaktů, musí dodávat částicím dispersní fáze kinetickou energii, dostačující k překonání energetické bariéry při přemístění částic ze stabilní polohy v prvním blízkém energetickém minimu za hranice druhého vzdáleného minima (podle Derjagina).
Možnost mezního rozpadu (destrukce) struktury při vibracích je potvrzena získáním úplných vibračně reologických křivek tečení řady vodních a nevodních vysoce koncentrovaných soustav, pro které se dosažení nejmenší úrovně newtonovské viskozity v nepřítomnosti vibrací, ukázalo nemožným (Obr. 5).
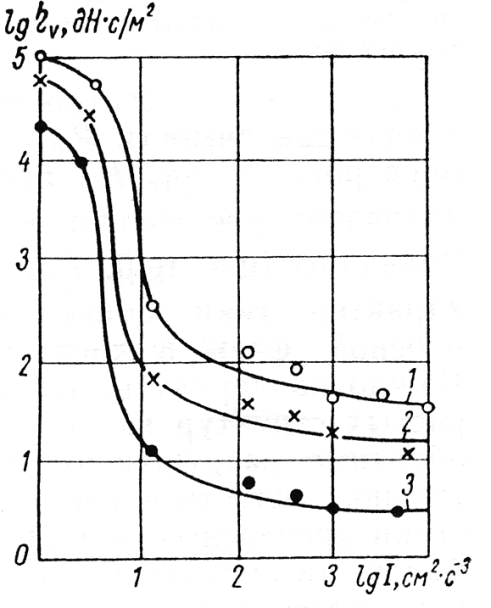
Současně je tato hladina rozpadu (destrukce) struktury dosažena při prudce narůstajícím výkonu vibračního pole I v oblasti ην→ ηm .
Analýza příčin tohoto jevu ukázala, že při destrukci (rozpadu) struktury v místech nejslabších kontaktů, vibrace současně zvyšuje pravděpodobnost srážek částic v lyofobních částech jejich povrchů, a to přispěním k překonání energetické bariéry mezi nimi, čímž dochází ke vzniku pevných kontaktů s fixací částic ve vzdálenosti blízké koagulaci.
Aby se odstranil jev svérázného vibračního zpevnění struktury v procesu její destrukce (rozpadu), vyvolávající nutnost prudkého zvýšení I v oblasti ην→ ηm je nutné vyloučit možnost tvorby pevných kontaktů s pomocí blokování povrchu částic adsorbovanými vrstvami PAL.
Skutečně, jak ukazují údaje uvedené na obr. 5, zavedení do vysoce koncentrované vodní disperse směsi hydrosilikátů a hydroaluminátů přísady ethoxylačního alkylfenolu (2) nebo methylsilikonátu sodíku (3) v množství vypočteném na základě podmínky vzniku nasycené adsorbované vrstvy, vede k největšímu relativnímu zmenšení výkonu vibračního pole, zejména v té oblasti mezní destrukce (rozpadu), kde je při absenci PAL nárůst výkonu největší. Současně s tím je efekt vibračního zpevnění struktury úplně odstraněn. Přitom stupeň největšího relativního poklesu výkonu vibrací (v oblasti ην = ηm obr. 5)
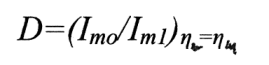
převyšuje 2 řády (5.102). Hodnotu Iml, odpovídající ηm1 je možné určit ze vztahu

Pokud je dynamický stav rozředěných koloidů charakterizován intenzitou Brownova pohybu dispersních fází, určeného energií tepelných kmitů molekul dispersního prostředí při stejných zbývajících podmínkách, pak v koncentrovaných mikroheterogenních dispersních soustavách je charakteristikou jejich dynamického stavu intenzita oscilací částic dispersních fází, určená výkonem vibračního pole zaváděného do soustavy I = a2 ω3.
Tudíž, veličina Dν= (I0 /I1 ) ην = ηm může posloužit dynamickým kriteriem při analýze zákonitostí kombinace působení PAL a vibračního pole v procesu rozpadu (destrukce) koagulačních struktur, tj. v dynamických podmínkách.
Nárůst veličiny D (obr. 5) ukazuje na jev vzájemného zesílení působení vibrací a adsorpčně aktivního prostředí, jež je charakterizováno podstatným (řádovým) relativním zmenšením výkonu vibračního pole za přítomnosti přísad PAL podle toho, jak dochází ke zvyšování stupně destrukce (rozpadu) struktury, tj. nárůstu fluidity soustavy.
Příčina vzájemného zesílení působení vibrací a adsorpčně aktivního prostředí v průběhu destrukce (rozpadu) koagulačních struktur spočívá v následujícím.
Vibrace, vedoucí k destrukci (rozpadu) struktury až do nejmenší hodnoty viskozity současně vedou ke vzniku pevných kontaktů. PAL, které jsou adsorbovány převážně na energeticky nejaktivnějších částech makro- mozaikového povrchu částic přirozeně oslabují strukturu jako celek, avšak co je ještě podstatnější, vylučují v procesu vibračního rozpadu (destrukce) možnost vzniku pevných kontaktů, odpovědných za růst výkonu vibračního pole v oblasti ην→ ηm. V důsledku toho v přítomnosti PAL výkon vibrací nejvíce klesá zejména v této oblasti. V tomto případě je třeba zvláště podtrhnout následující skutečnost.
Použití vibrací ke snížení viskozity směsí a k formování výrobků, stejně jako zavedení plastifikujících přísad do směsi je v současné technologii polymerbetonových směsí natolik akceptovanou skutečností, že by nebylo nutným dokazovat účelnost použití těchto metod také při vypracování technologie koloidních malt.
Nicméně, nutno brát v úvahu, že pouze ta společná kombinace vibrací a přísad PAL, při níž je dosahována nejnižší úroveň viskozity směsí při minimálních energetických ztrátách, odpovídá optimálním parametrům technologie přípravy dispersních materiálů, včetně koloidních malt. Tato, pro technologii nejdůležitější okolnost se zpravidla nerespektovala, což vysvětluje neexistenci výzkumů orientovaných na problematiku společného působení vibrací a PAL v technologii přípravy polymerbetonových směsí. Mezitím právě optimální kombinace těchto činitelů, což plyne ze samotné podstaty vysoce koncentrovaných dispersních soustav, by měla být základem pro určování parametrů přípravy, nanášení a hutnění (tvarování) směsí s nízkým obsahem kapaliny.
4. Základní etapy vytváření struktur
K efektivnímu řízenu procesu vytváření dispersních materiálů a pevných látek se zadanou strukturou a vlastnostmi je nutné zkoumat zákonitosti vytváření struktury ve všech etapách, počínaje momentem interakce pevné a kapalné dispersní fáze až po ukončení fázových přechodů v soustavě.
Řízení procesu vytváření struktur je nejefektivnějším v jeho počátečných stádiích, v období, kdy v soustavách převažují struktury koagulačního typu. Vlastnosti struktury zejména v této etapě v podstatě určují hustotu, homogenitu, a stupeň disperse výsledné struktury reálných pevných látek a dispersních materiálů po ukončení fázových přechodů (krystalizace, polymerace).
Dříve bylo ukázáno, že optimální forma působení na soustavu, při směšování dispersních fází, zhutnění soustav a jejich deformaci, v období převážně koagulačního vytváření struktury, spočívá ve vibračním působení v kombinaci s malými přísadami povrchově aktivních látek.
K určení zákonitostí vytváření vysoce zaplněných dispersní pevnou fází tixotropních koagulačních struktur byly zkoumány zvláštnosti změn makrostruktury při vibračním směšování makrostruktury modelové soustavy, vytvořené dispersí přírodního vápníkového bentonitu (CaB) v kombinaci s jemně dispersním (S=3000 cm2/g) křemenem: poměr hmotností CaB:Si02= 20:80, poměr voda pevná fáze B/T= 0,19 — 0,25.
Převažující význam směšování ve srovnání se jinými technologickými postupy je dán tím, že základy budoucí struktury se tvoří již v procesu vzájemné distribuce složek, které jí tvoří.
Tabulka 3.
Kinetika změn strukturních charakteristik soustavy v procesu vibrace
| Doba míchání, s. | Hustota agregátů, g/cm2 | Střední specifický povrch agregátů, cm2/g | Mezní napětí ve smyku, Pa.10-3 | Střední vypočtená kohezní síla v kontaktu Fc, 1012N |
|---|---|---|---|---|
| 10 | 1,50 | 4000 | 200 | 7,8 |
| 20 | 500 | 5,8 | ||
| 30 | 1,66 | 220 | 1000 | 6,5 |
| 45 | 150 | 3000 | 9,7 | |
| 60 | - | 110 | 4000 | 9,8 |
| 90 | 1,74 | 100 | - | - |
| 120 | 1,76 | 90 | 5000 | 9,2 |
| 180 | 1,85 | - | 6000 | 86 |
| 360 | 1,90 | 75 | 7000 | 47 |
| 600 | 1,91 | 74 | 6900 | 60 |
Kinetika vytváření struktury byla zkoumána v procesu smíchávání výchozí pevné a kapalné fáze ve speciálně navrženém lopatkovém vibračním směšovači o kapacitě 2000 cm3 s regulovatelným počtem otáček ( od 14 do 220 otáček za minutu), parametry vibrací tělesa směšovače s proměnnými frekvencemi (500 — 2200 min.-1) a amplitudami (0,5 — 2 mm).
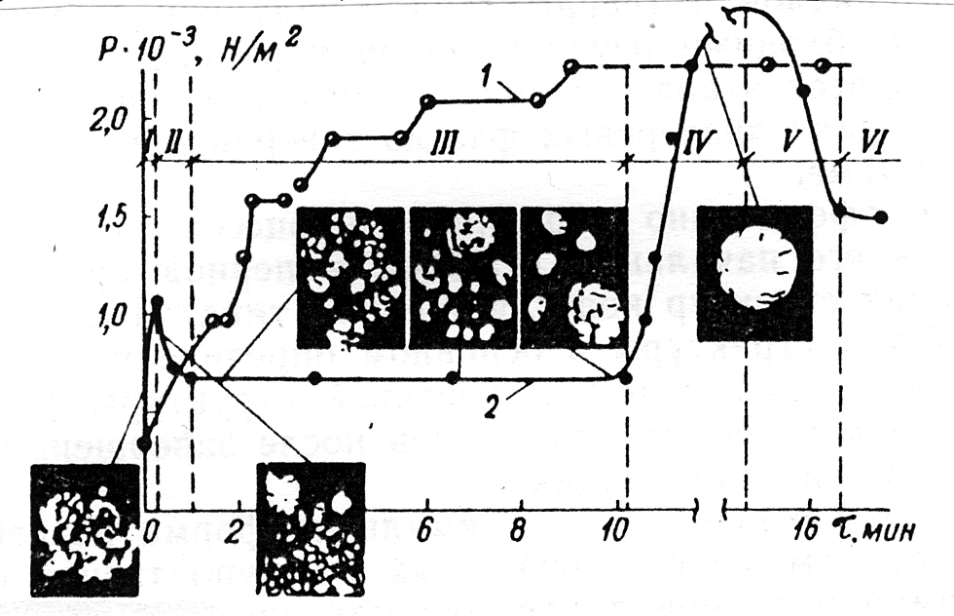
Studium kinetiky změny těchto veličin, především η, P(τ), ukazuje na podstatné kvalitativní rozdíly v procesech vytváření struktury bez vibrací a v kombinaci s nimi (Obr.6, Tab. 3).
Na počátku procesu vibračního směšování probíhá vytváření a destrukce (rozpad) agregátů tvořených jemnými dispersními částicemi pevné fáze (etapy I a II). Po dosažení prvního maxima η; P(τ) se převládajícím stává proces destrukce (rozpadu) struktury na jednotlivé houbovité agregáty. Prvky struktury houbovitého agregátu jsou smáčené kapilární menisky mezi jednotlivými skupinami částic pevné fáze spojené kapilárním tlakem Pσ, jehož maximální hodnota odpovídá úplnému smáčení Pσ= 4σ/δ, kde σ je povrchové napětí na rozhraní kapalina — pára a δ rozměr jednotlivých agregátů tvořených částicemi pevné fáze.
Tím se vysvětluje poměrně snadná destrukce (rozpad) velkých houbovitých agregátů s vytvářením agregátů drobnějších a pevnějších.
Souběžně se vznikem smáčených menisků, vytvořením hrubě dispersní struktury, houbovitých agregátů z ní vzniklých a jejich destrukcí (rozpadem), probíhá nepřetržitá migrace kapalné fáze pod vlivem kapilárního tlaku do nejužších mezer mezi částicemi. Tento proces pokračuje i tehdy, když destrukce (rozpad) houbovitých agregátů je u konce a vznikly z nich drobnější a hutnější agregáty ve formě granulí. Uvnitř granulí se ustálil takový stupeň mikro- makro- nehomogennosti struktury a rozdělení různých pevných fází, který koresponduje stavu soustavy na konci předchozí etapy (Obr.7).
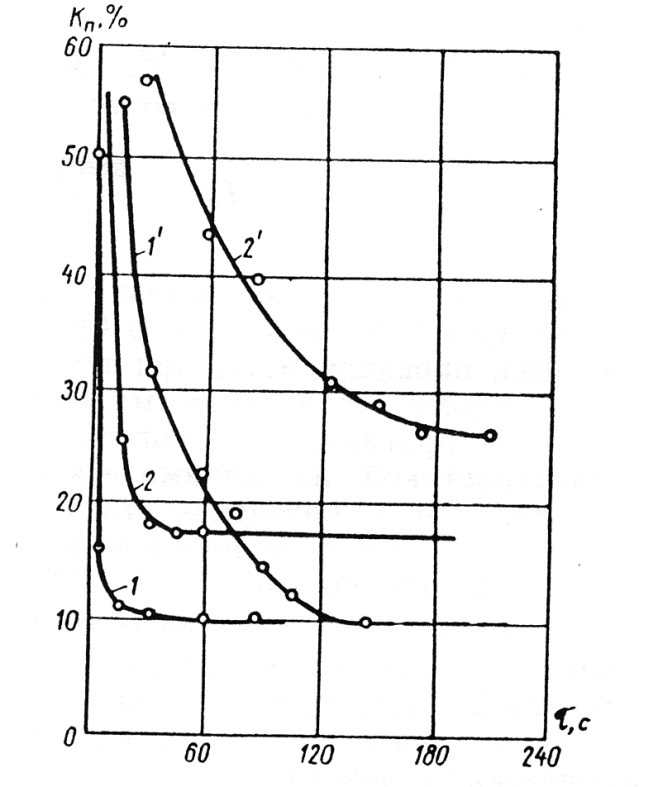
prášcích (1, 2) a v soustavě s kapalným dispersním prostředím (1' 2´) při míchání s vibrací (1, 1´) a bez ní (2, 2´)
Konstantnost hodnot η; P(τ) v III etapě ukazuje, že v makrostruktuře soustavy s mícháním jako celku nedochází k podstatným změnám. Avšak uvnitř agregátů přitom pokračují aktivní procesy objemové migrace do nejtenčích pórů a k dvourozměrné migraci kapalné fáze směrem k oblastem skutečných kontaktů mezi částicemi.
Tato etapa je charakterizována převažující rolí procesu dvourozměrné migrace. Je doprovázena vznikem dvourozměrného tlaku podél rozhraní adsorpčních vrstev kapaliny, dostačujícímu k tornu, aby odsunul skutečné kontakty atomů do vzdálenosti větší nebo rovné vzdálenosti blízké koagulaci (10-9cm):

kde b=RTΓm , κ =Γ/Γm — adsorpční pokrytí (κ< 1).
Přitom dochází ke snížení pevnosti kontaktu (adsorpční snížení pevnosti), nicméně celkově pevnost struktury agregátu vzrůstá proto, že vibrace snadno zvyšuje hustotu uspořádání částic v agregátu (nárůst φ) a tudíž snížení pevnosti kontaktu je kompenzováno zvýšením počtu částic v objemové jednotce.
V dalším v důsledku intensivních nárazů agregátů v průběhu vibrací, počet skutečných koagulačních kontaktů s fixací částic pevné fáze ve vzdálenosti blízké koagulaci uvnitř agregátu roste, což se potvrzuje růstem vypočtené veličiny Fc (Tab. 3).
Vznik takových koagulačních kontaktů v celém objemu granulí, plná smáčivost všech vnitřních povrchů částic v agregátech — granulích je v podstatě nejvýraznější charakteristická zvláštnost této etapy, jíž je proto možné nazvat etapou formování souvislé koagulační struktury uvnitř agregátů.
Závěrečná etapa vytváření struktury je koalescence agregátů — granulí. Tomuto procesu napomáhají i aktivní nárazy při vibracích v kombinaci se smykovými deformacemi celé soustavy v důsledku otáčení lopatek směšovače. V této etapě (IV) převládá vytvoření souvislé plasticko-viskózní (nebo pružně-plasticko-viskózní) soustavy nad procesy její destrukce (rozpadu), přičemž smykové efektivní napětí P(τ) a viskozita prudce narůstají. Vzniklá souvislá dvoufázová koagulační struktura je natolik pevná, že její rozpad (destrukce), při dané konstantní intenzitě mechanického působení, může být nepatrným, vzdáleným od mezního (etapy V a VI). Takovým způsobem při přechodu od jedné etapy k následujícím vytvářejí se stále hutnější a pevnější prvky struktury. Současně s tím stupeň nerovnoměrnosti distribuce částic různých druhů pevné fáze zafixován v předchozí etapě dokonce i při dlouhodobém působení na soustavu se předává „ jako dědictví" do následujících etap, v nichž je dosažení homogennosti distribuce ještě více ztíženo vznikem pevnějších strukturních prvků. Proto k dosažení maximální homogenity a hustoty v závěrečné etapě koagulačního vytváření struktur je nutné zajistit mezní destrukci toho typu struktury, který je základním v každé z předchozích etap vytváření struktur.
Tyto přechody z jednoho typu makrostruktury k druhému mohou být dovršeny při vibračním směšování a to částečně nebo úplně. V soustavách vysoce naplněných dispergovanou pevnou fází je přechod k souvislé hutné dvoufázové koagulační struktuře možný pouze při následném vibračním zhutnění doprovázeném deformací soustavy.
Tudíž při vibracích o specifickém výkonu Ic=α2 ω3, odpovídajícímu hranici přechodu od pseudozkapalnění k pseudotvaru, proces vytváření struktury se dělí na postupné etapy lokalizované v celém objemu a v čase. Důležitá odlišnost strukturovaných soustav s vysoce dispersní pevnou fází od hrubě dispersních spočívá v tom, že pro první je tato hranice určena konstantní hodnotou specifického výkonu I a pro druhé je dána konstantní veličinou zrychlení αω2/g ≈ 1.
Pro I = Ic pravděpodobnost homogenní distribuce gradientu rychlostí v celém objemu soustavy je maximální. Důsledkem toho je prostorově časová homogennost při destrukci (rozpadu) a vytváření struktury a v distribuci rozličných dispersních fází, což určuje homogennost soustavy jako celku. Současně s tím prostorově časová homogennost při destrukci (rozpadu) a vytváření struktury je základní příčinou rozdělení procesu vytváření struktury jako celku na jednotlivé etapy.
Volba optimálních parametrů technologických procesů zpracování strukturovaných dispersních soustav a získávání dispersních materiálů musí být uskutečňována v souladu se základními etapami koagulačního vytváření struktur a režimy technologických operací, mezi něž patří intensita a délka směšování, zhutňování a formování, musí odpovídat optimální úrovni destrukce struktury v každé etapě vytváření struktury. Současně s tím je možné oddělení procesu transportu hmoty v čase od procesů aktivních fázových transformací v celém objemu soustavy.
5 Specifika reologických vlastností strukturovaných dispersních soustav
V souladu s principy fyzikálně chemické mechaniky nejdůležitější podmínkou získání materiálů s danými strukturně mechanickými vlastnostmi je řízení dispersních soustav ve všech etapách procesu vytváření struktury. Zvláštní úlohu při zabezpečení potřebné struktury (pórovitost, rozměr, tvar porézních kapilár, rozměr krystalů v krystalické srostlici atd.) a nezbytných vlastností materiálu ( pevnost, moduly deformace, životnost, vodotěsnost atd.), má řízení procesu vytváření struktury v počáteční etapě.
S cílem vědecky zdůvodněného výběru optimálních parametrů působení na soustavu v počáteční etapě využívá fyzikálně chemická mechanika řadu metod měření strukturně mechanických reologických charakteristik, dovolujících všestranně zhodnotit pevnostní vlastnosti vznikajících struktur a tudíž i možnost a nutnost jejich řízení.
Mezi různorodými typy viskózních kapalin vyčleňujeme, podle reologických vlastností v stacionárním laminárním toku, dvě základní třídy kapalin: 1) Newtonovské kapaliny, jejichž viskozita nezávisí na působícím na ně napětí smyku a na gradientu rychlosti deformace; 2) Strukturované dispersní soustavy, u nichž veličina efektivní viskozity se může měnit v širokém rozmezí v závislosti na působícím napětí smyku a gradientu rychlosti. Takovými strukturovanými soustavami jsou koagulační struktury.
V počátečním období, charakterizovaném přítomností kapalné a pevné fáze, obsahuje v pevné fázi alespoň nepatrné množství částic koloidních rozměrů. V tomto případě vzniká prostorová strukturní mřížka, jejíž pevnost je určena vzdáleností mezi uzly a tloušťkou mezivrstvy dispersního prostředí a která má stínící účinek na molekulární sily adheze mezi částicemi. Míra stínění závisí na tloušťce mezivrstev, která je návazně závislá na počtu a rozměrech koloidních částic v jednotkovém objemu. Čím víc částic je rozmístěno v objemu, tím slabší je stínící působení mezivrstvy dispersního prostředí mezi částicemi prostorové mřížky, tím bližší je vzdálenost mezi uzly prostorové mřížky a tudíž tím pevnější je struktura.
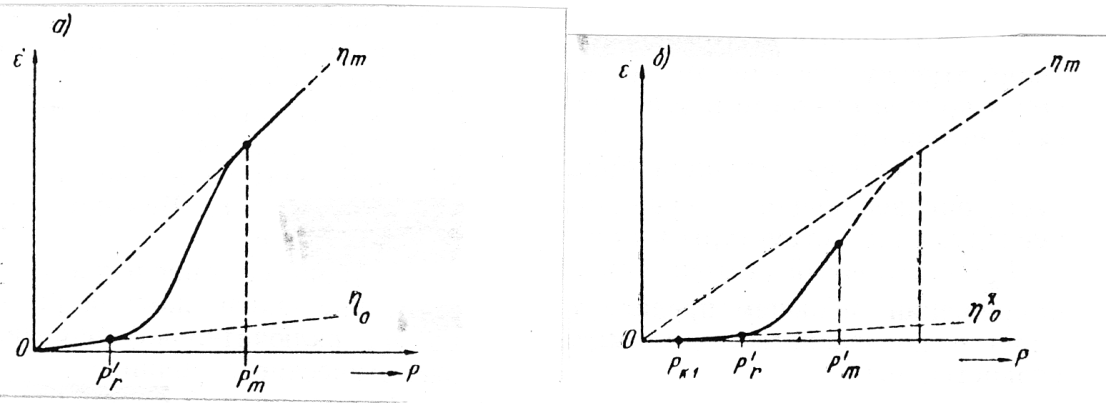
Jednou ze zvláštností koagulační struktury je schopnost obnovovat v klidu, částečně do parametrů rovnovážného stavu při daném gradientu rychlosti, rozpadlou při pohybu, prostorovou mřížku na účet tepelného (Brownova) pohybu částic s koloidními rozměry (tixotropie). Proto strukturované kapaliny, na rozdíl od skutečných newtonovských, mají proměnnou viskozitu v závislosti na rychlosti deformace (Obr. 8).
Tudíž koagulační struktury representují pružně viskózně plastická tělesa, u nichž pozorujeme mez fluidity a při velmi malých napětích smyku projevují vlastnosti tečení s konstantní maximální viskozitou ηo. S růstem napětí smyku stupeň rozpadu struktury narůstá, při plném rozpadu je struktura charakterizována minimální viskozitou ηm, která již nezávisí na smykovém napětí a gradientu rychlosti v podmínkách stacionárního toku. Viskozita, která může být pozorována v intervalu ηm < η < ηo se nazývá efektivní viskozitou ηp¬ .Viskozita stanovená při vibracích je vibrační viskozitou ην .
V průběhu stanovení charakteristických reologických konstant při pořízení úplné reologické křivky ( počínaje s ηo do ηm včetně) se stanovují taktéž některé základní reologické veličiny:
- Pk2 smluvená Bingamova mez fluidity, odpovídající úsečce vymezené průsečíkem tangenty ke křivce viskozity v zóně prakticky konstantní plastické viskozity s osou napětí smyku ;
- Pr —smluvená hranice prakticky nerozpadlé struktury, určující v podstatě hranici konstantní viskozity neporušené struktury v dyn/cm2;
- Pkl smluvená statistická mez fluidity v dyn/cm2; při napětích menších než je mez fluidity vzniká pružnost, dopružování a zpomalené tečení s maximální viskozitou ηo ;
- PΓ — smluvená hranice mezně rozpadlé struktury v dyn/cm2;
- Pm — pevnost struktury při pružně křehkém nebo elastickém přetržení v dyn/cm2 nebo kg/cm2.
Pro strukturované soustavy existuje pouze určitá oblast minimálně možných hodnot viskozity ηm ,odpovídající určitým hodnotám gradientu rychlosti a napětí smyku. S dalším zvyšováním napětí smyku a gradientu rychlosti takové soustavy přecházejí do turbulentního stavu, v němž viskozita narůstá.
Pro výzkum reologických charakteristik dispersních soustav se používají viskozimetry různých typů, které umožňují stanovit viskozitu, mez pevnosti a další vlastnosti soustav.
Nejúplnější charakteristiku reologických vlastností strukturovaných dispersních soustav je možné získat při použití rotačních viskozimetrů, u nichž je zkoumaná soustava umístěna v úzké mezeře mezi koaxiálními válci. Při otáčení vnitřního válce' vzhledem k nepohyblivému (vnějšímu) ve zkoumaném materiálu rozmístěném v úzké mezeře mezi válci dochází k posuvu jedné vrstvy vzhledem ke druhé (minimální tloušťka elementární vrstvy je přibližně dána středním průměrem částic). Maximální rychlost posuvu vzniká ve vrstvě, která je v kontaktu s vnitřním válcem, na povrchu vnějšího válce je rychlost rovna nule. Spád rychlosti podél tloušťky vrstvy materiálu, který může být charakterizován veličinou gradientu rychlosti έ= dν/dh, kde ν je lineární rychlost a h mezera, je zadán rychlostí otáčení vnitřního válce a závisí na poloměru válců a velikosti mezery.
Gradient rychlostí v úzké mezeře rotačního viskozimetru se stanovuje podle vztahu
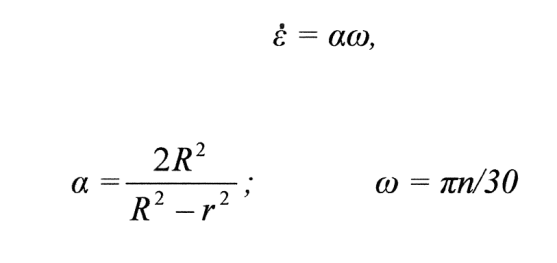
έ je gradient rychlostí v s-1 , R a r poloměry vnějšího, potažmo vnitřního válce v cm, n počet otáček vnitřního válce v ot/min a ω úhlová rychlost v rad/s.
Rotační elektronově elastoviskozimetry umožňuí měnit gradienty rychlosti v toku v širokých mezích ( s rozdílem 107 krát). V procesu měření se stanovují závislosti gradientu rychlostí έ ve stacionárním toku na smykových napětích P při konstantní rychlosti deformací pro každé zaměření. Momenty, odpovídající smykovým napětím, vznikající při otáčení válce jsou zapisovány oscilografem. Díky možnosti měnit gradient rychlostí v širokém rozsahu, lze získat úplnou charakteristiku reologických vlastností soustav.